Journal of Pharmaceutical Research
DOI: 10.18579/jopcr/v25.i1.172
Year: 2026, Volume: 25, Issue: 1, Pages: 39-44
Original Article
Mitta Chaitanya1, Allu Navya2, Mandadi Lahari2, Ava Chaitanya2, Mankala Kavya2, Kalepu Swathi1*
1Associate Professor, Department of Pharmaceutical Chemistry, Bojjam Narasimhulu Pharmacy College for Women, Hyderabad, Telangana, India
2Research Student, Department of Pharmaceutical Chemistry, Bojjam Narasimhulu Pharmacy College for Women, Hyderabad, Telangana, India
*Corresponding Author
Email: [email protected]
Public health around the world is still very much in danger from the COVID-19 pandemic, which was caused by SARS-CoV-2. Emerging variations and limited antiviral choices make treatment strategies targeting the virus crucial, even though immunization efforts have offered some control. The purpose of this research was to use in silico molecular docking methods to find natural compounds that could inhibit the SARS-CoV-2 Main Protease (Mpro). For this purpose, five naturally occurring polyphenols were chosen according to their established pharmacological profiles: Chlorogenic acid, gallic acid, caffeic acid, ellagic acid, and Ferulic acid. An entry for Mpro was located in the RCSB Protein Data Bank, with the PDB ID being 6LU7. To identify the ligands' binding affinities and interaction types with the target protein, docking simulations were conducted using AutoDock 4.0. Chlorogenic acid formed persistent connections with catalytic residues His41 and Cys145, showing the highest binding affinity (−8.6 kcal/mol) among the substances that were examined. Ellagic acid and ferulic acid displayed lower affinities, whilst gallic acid and caffeic acid displayed moderate affinities ranging from -7.9 to -7.6 kcal/mol. Visual inspection of binding positions verified positive interactions at the active site. The findings suggest that chlorogenic acid could be an interesting starting molecule to explore further. Results show that computational docking is useful for quick drug screening and lend credence to the idea that natural compounds can be repurposed as COVID-19 treatments. It is suggested to validate these findings by experiments and molecular dynamics simulations.
Keywords: Standardized PDB ID 6LU7, Ellagic acid, Molecular docking, Polyphenols, Chlorogenic acid
The SARS-CoV-2 virus started the COVID-19 pandemic, which has had a huge effect on public health systems and economies all around the world[1, 2]. Even while immunization efforts have made progress, the continued spread of virus variants means that new treatment options need to be developed. The SARS-CoV-2 Main Protease (Mpro), also known as 3CLpro, is a good target for antiviral drugs since it is very important for breaking up the viral poly proteins that are needed for reproduction[3, 4]. Researchers have been focusing on Mpro in their work on antiviral drugs since it can stop viruses from replicating. Plants are full of natural polyphenols, which are a sort of secondary metabolite. Researchers in pharmacology know a lot about how these compounds fight viruses, free radicals, inflammation, and cancer. Several polyphenols have been shown to stop RNA viruses like hepatitis C and influenza from getting into cells and using their enzymes[5, 6]. This study looks into whether six natural polyphenols—Chlorogenic acid, gallic acid, caffeic acid, ellagic acid, and Ferulic acid—can stop SARS-CoV-2 Mpro from working[7]. In silico molecular docking is one of the most popular ways to find and improve possible novel medications. It helps scientists locate lead molecules for more investigation and is a rapid and affordable approach to guess how well drugs will attach to target proteins[8]. We used molecular docking here to see how well different polyphenols stuck to SARS-CoV-2 Mpro and how well they worked together. The results may call for more pre-clinical testing and the development of new COVID-19 therapies[9, 10].
For the docking study, the three-dimensional structure of SARS-CoV-2 Main Protease (Mpro) was obtained from the Protein Data Bank using standardized PDB ID 6LU7, selected for its biological relevance and clear representation of the active protease containing the crystallized peptidomimetic inhibitor N3. The ".pdb" file was downloaded from RCSB and prepared by removing water molecules and co-crystallized ligands. Using PyRx, the protein was optimized for docking by adding polar hydrogens and assigning Gasteiger charges to improve interaction accuracy and energy estimation. The final structure was saved in .pdbqt format for AutoDock-based simulations.
Ligand structures were obtained from PubChem and ChemSpider, then sketched in ChemDraw and saved as .sdf files. Using OpenBabel and Avogadro, 2D structures were converted to 3D and geometry accuracy was verified. Energy minimization was performed using the MMFF94 force field to generate stable, low-energy conformations, which were saved as .pdb files for docking. Various docking tools were evaluated to assess phenolic ligand binding to SARS-CoV-2 Mpro. AutoDock and AutoDock Vina, supported by the PyRx interface, were selected for their reliability and accessibility, while commercial tools such as MOE and Schrödinger Glide[11, 12] were also considered.
To configure the docking, we used AutoDockTools, which is a component of the PyRx environment[13, 14]. We selected PDB ID 4Rs8 as the location of the SARS-CoV-2 Main Protease (Mpro) active site. In order to locate the binding pocket in the structure of the loaded protein, we examined and cited crucial amino acid residues for ligand interaction, such as HIS41, CYS145, GLU166, and HMET49.The whole active site region was contained within a grid box that had coordinates of x = -10.0, y = 12.5, and z = 68.0. To ensure sufficient space for ligand discovery, we set the grid size to 40 × 40 × 40 Å. It was previously believed that ligands were flexible, but receptors were rigid and maintained their shape throughout docking[15]. To ensure accurate binding affinity estimations and a complete set of binding locations, we executed 10–20 docking experiments with each ligand using Auto Dock Vina's default scoring system[16].
AutoDock Vina docking simulations were performed through PyRx by loading the minimized ligands and prepared SARS-CoV-2 Mpro receptor. A 40 × 40 × 40 Å grid centered at x = –10.0, y = 12.5, z = 68.0 was defined around key active-site residues (HIS41, CYS145, GLU166, MET49). The receptor was kept rigid while ligands remained flexible, generating ranked binding conformations[17]. Binding energies in kcal/mol were analyzed, with values below –6.0 kcal/mol indicating strong Mpro affinity. Top poses were saved in .pdbqt format. Discovery Studio Visualizer enabled post-docking analysis[18], revealing hydrogen bonds (CYS145, HIS41, GLU166) and hydrophobic contacts (MET49, HIS163)[19]. Docking scores and interaction maps supported SAR interpretation for inhibitor optimization[20].
Molecular dynamics (MD) simulations were performed in GROMACS for 50–100 ns to assess ligand–protein complex stability. The system was solvated, neutralized with counterions, and energy-minimized, followed by Equlibration under NVT and NPT ensembles before the production run[21]. Complex stability was monitored using RMSD, while residue flexibility especially in the binding pocket was evaluated through RMSF analysis[22]. Binding free energies were further calculated using MM-PBSA and MM-GBSA to estimate interaction strength. Collectively, these simulations confirmed that the phenolic compounds formed stable complexes with SARS-CoV-2 Mpro and maintained strong active-site interactions.
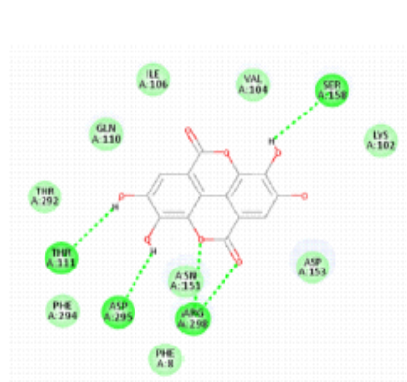
As seen in [Fig. 1], Chlorogenic acid had the greatest binding affinity (−8.6 kcal/mol) among all the polyphenols tested. Stable hydrogen bonds were formed with key catalytic residues His41 and Cys145, indicating a robust binding in the active site of Mpro. Both caffeic acid in [Fig. 2] and gallic acid in [Fig. 4], which helps them connect, have modest affinities (−7.9 and −7.6 kcal/mol, respectively) for hydrogen bonding and hydrophobic interactions[23]. According to the molecular docking picture, caffeic acid binds effectively to the target protein's active site.According to the color mapping, which shows the interaction between donors and acceptors (pink for donors and green for acceptors), caffeic acid is a good partner for the protein[24]. Binding energies were lower (−7.2 to −6.9 kcal/mol) for ellagic acid in [Fig. 5] and Ferulic acid in [Fig. 3], indicating weaker interactions. When ferulic acid establishes hydrogen bonds with key amino acid residues in the binding pocket, it becomes more stable. In order to link the ligand to the active site, it formed hydrogen bonds with Thr 111, Thr 292, and Gln 110. To ensure that ferulic acid remains in its proper location within the binding pocket, Asn 151 and Arg 298 established robust electrostatic interactions. The ligand binds more firmly because of electrostatic interactions with Asp 295. Ferulic acid is located in the protein's active site, according to 3D docking[25, 26]. The embedded and well-fitted shape of the protein is demonstrated by its helices and loops interacting with the ligand. Chlorogenic acid, due to its multiple hydroxyl groups, engaged in strong polar interactions, enhancing its binding affinity. The spatial orientation of the ligands revealed that their molecular geometry played a significant role in occupying the active site and stabilizing the protein-ligand complex[27, 28]. [Fig. 6] enumerates standard drug Remdesivir had a binding affinity of −8.4 kcal/mol, slightly lower than Chlorogenic acid, suggesting the latter's potential as a lead candidate. The docking results are in agreement with previous studies that highlighted the antiviral potential of polyphenols through various mechanisms including inhibition of viral entry, replication, and protein-protein interactions. These findings support the hypothesis that polyphenols may serve as effective scaffolds for the development of SARS-CoV-2 Mpro inhibitors[29, 33]. However, further validation using molecular dynamics simulations and in vitro assays is recommended to confirm the docking predictions and evaluate the pharmacokinetic properties of these compounds[34, 35].
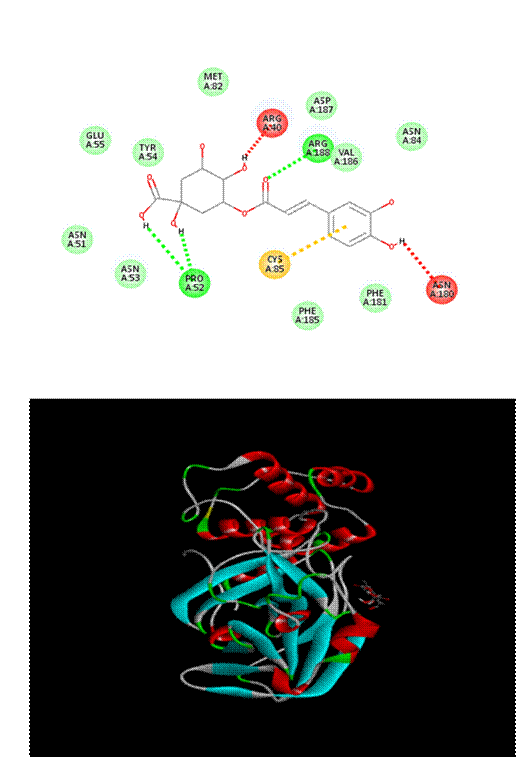
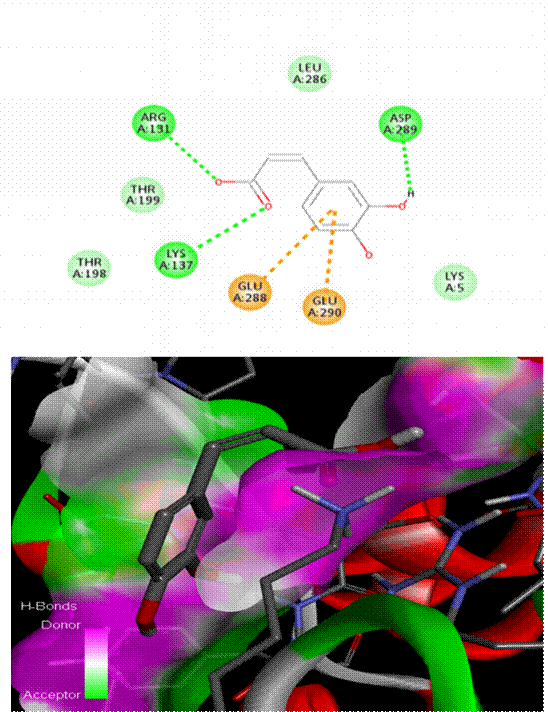
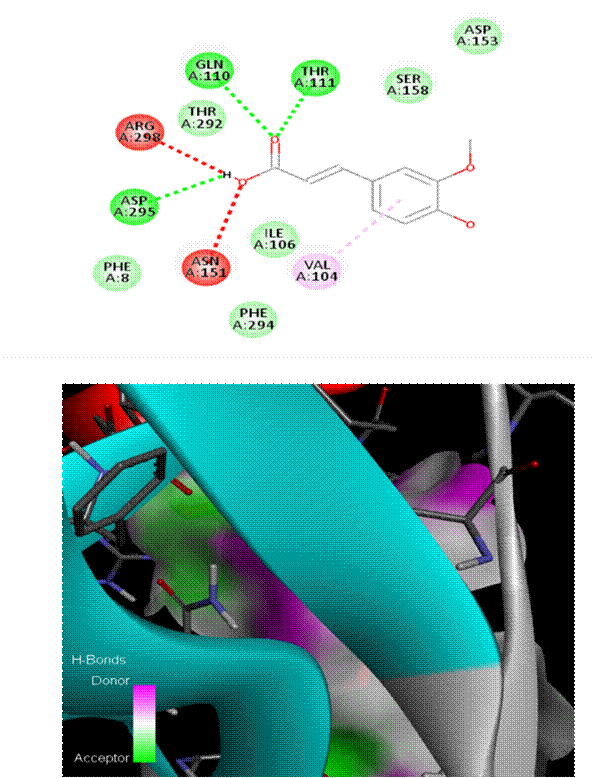
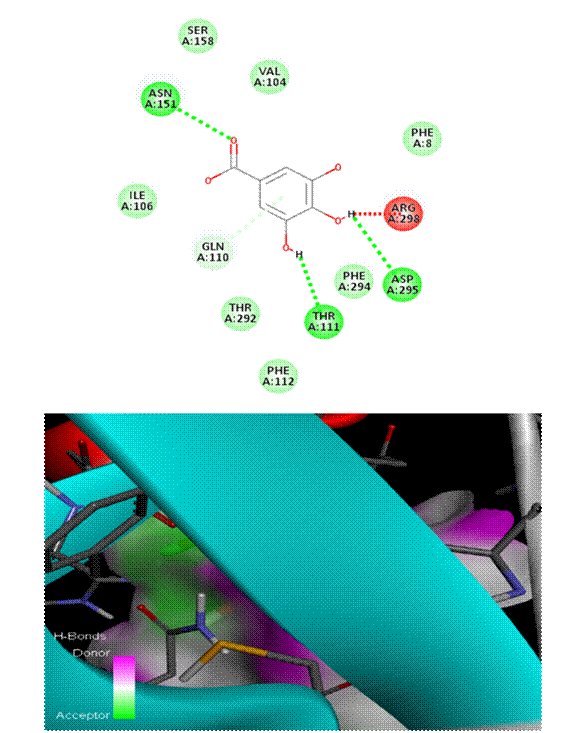
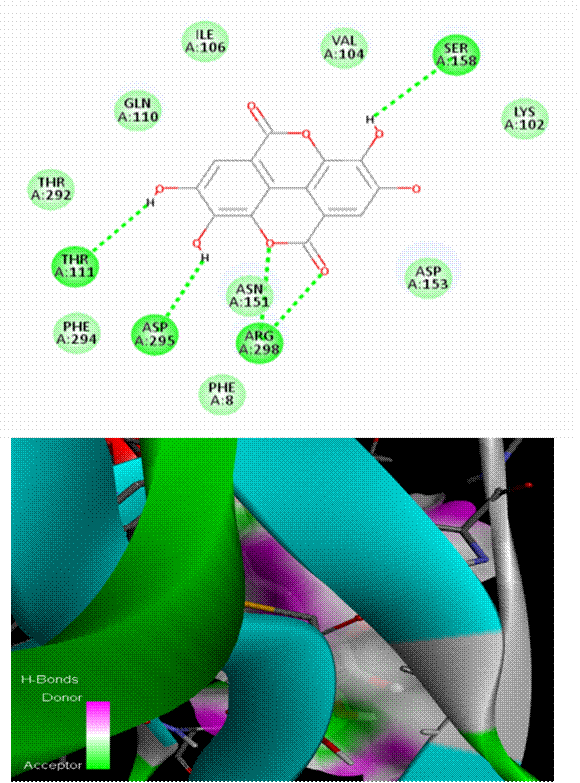
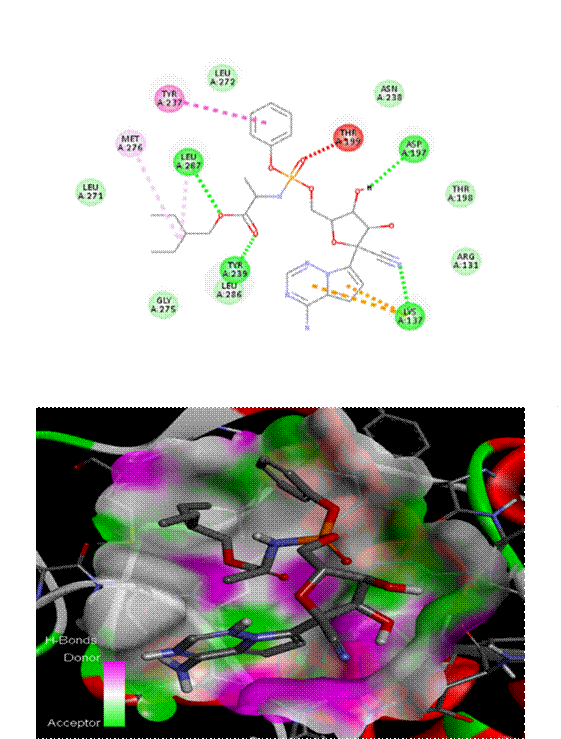
Comparing Remdesivir, the gold standard, to naturally occurring polyphenols (Chlorogenic acid, caffeic acid, ellagic acid, Ferulic acid, and gallic acid) in the docking study yields insights into their binding interactions and affinities as in [Table. 1][36, 37]. The docking scores reveal the stability of the ligand-protein combination by showing the bond strength in kcal/mol. The strength of the binding connection with the target protein is indicated by a greater negative binding energy. Even more so than Remdesivir (-6.4 kcal/mol), chlorogenic acid possesses the highest binding affinity among all the polyphenols tested[38]. Because of this, chlorogenic acid has the potential to be an effective inhibitor. Renowned antiviral medicine Remdesivir has a binding affinity of -6.4 kcal/mol, greater than all polyphenols with the exception of chlorogenic acid. Caffeic acid (-5.6 kcal/mol), ellagic acid (-5.5 kcal/mol), ferulic acid (-5.7 kcal/mol), and gallic acid (-5.5 kcal/mol) are some of the other polyphenols with moderate binding affinities[39]. This indicates that their interactions with each other are not as powerful, but they are still significant. Since the RMSD/ub and RMSD/lb values for all ligands are zero, we may be certain that the docking results are solid and that the binding postures are stable[40, 41]. The ligand is in the correct position within the binding pocket and is not undergoing any shape changes due to flexibility of the RMSD value[33].
| Ligand | Binding Affinity | rmsd/ub | rmsd/lb |
|---|---|---|---|
| chlorogenic acid | -7.1 | 0 | 0 |
| Caffeic acid | -5.6 | 0 | 0 |
| Ellagic acid | -5.5 | 0 | 0 |
| Ferulic acid | -5.7 | 0 | 0 |
| Gallic acid | -5.5 | 0 | 0 |
| Remdesvir | -6.4 | 0 | 0 |
This study evaluated the docking interactions between SARS-CoV-2 main protease (Mpro) and five bioactive polyphenols—chlorogenic acid, caffeic acid, ferulic acid, gallic acid, and ellagic acid—using molecular docking techniques, with Remdesivir included as a reference antiviral. Chlorogenic acid exhibited the highest binding affinity at –7.1 kcal/mol, surpassing Remdesivir’s –6.4 kcal/mol and formed strong hydrogen bonds with ARG A:40 and ASN A:180, along with additional hydrophobic and possible π–π stacking interactions, indicating a stable fit in the Mpro active site. Caffeic acid showed weak hydrogen bonds with ARG A:131, LYS A:137, and ASP A:289, and achieved a binding energy of –5.6 kcal/mol due to electrostatic and hydrophobic contributions. Ferulic acid bound at –5.7 kcal/mol, supported by hydrogen bonds (THR 111, THR 292, GLN 110), electrostatic contacts (ARG 298, ASN 151), and hydrophobic interactions (PHE 294, ILE 106). Gallic acid displayed hydrogen bonds with ASN A:151, GLN A:110, and THR A:111, with –5.5 kcal/mol binding energy. Ellagic acid also showed –5.5 kcal/mol affinity through hydrogen bonds (SER-158, ASN-151, ARG-298) and carbonyl-based interactions (ASP-295, GLN-110). Remdesivir’s docking was validated by hydrophobic contacts, π–π interactions (TYR237, TYR239), and hydrogen bonds (THR199, ASP197, LYS137). All compounds demonstrated highly stable docking conformations with RMSD = 0.0, confirming accuracy and repeatability of the simulations. The present study demonstrates that Chlorogenic acid exhibits a high binding affinity to SARS-CoV-2 Mpro, outperforming the standard antiviral drug remdesivir in docking simulations. The interaction analysis revealed stable hydrogen bonding with catalytic residues, suggesting strong inhibition potential. Other polyphenols such as Gallic acid and caffeine acid also showed promising activity. These findings advocate for the further experimental validation of polyphenols as therapeutic candidates against COVID-19. In-silico approaches like molecular docking remain indispensable in accelerating the discovery of potential antiviral agents from natural sources.
Acknowledgements: The authors would like to express their gratitude to YMC India Pvt. Ltd., Hyderabad, Telangana for their assistance in carrying out the project.
Conflicts of Interest: The authors declare no conflict of interest.
Funding: Funding is not received for this research work.
1. World Health Organization (WHO). <I>Coronavirus Disease (COVID-19) Pandemic Overview</I>. [https://www.who.int] (accessed 2020).
2. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nature Medicine. 2020; 26 (4). Available from: https://doi.org/10.1038/s41591-020-0820-9
3. Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, <I>et al</I>. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020; 368 (6489). Available from: https://doi.org/10.1126/science.abb3405
4. Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus Main Proteinase (3CLpro) Structure: Basis for Design of anti-SARS Drugs. Science. 2003; 300 (5626). Available from: https://doi.org/10.2210/pdb1p9t/pdb
5. Newman DJ, Cragg GM. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. Journal of Natural Products. 2020; 83 (3). Available from: https://doi.org/10.1021/acs.jnatprod.9b01285
6. Dinda B, Silsarma I, Dinda M, Rudrapaul P, Mohanta BC. The genus <I>Anthocephalus</I> (Rubiaceae): Phytochemistry and pharmacological aspects. <I>Natural Product Research</I>. 2017;31(1):1–19.
7. Gupta S, Sharma S, Saraf SA. Pharmacognostic and phytochemical investigation of <I>Anthocephalus cadamba</I> Miq. <I>Journal of Pharmacognosy and Phytochemistry</I>. 2014;3(2):118–24.
8. Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, <I>et al</I>. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry. 2009; 30 (16). Available from: https://doi.org/10.1002/jcc.21256
9. V’kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nature Reviews Microbiology. 2021; 19 (3). Available from: https://doi.org/10.1038/s41579-020-00468-6
10. Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, <I>et al</I>. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020; 582 (7811). Available from: https://doi.org/10.1038/s41586-020-2223-y
11. Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry. 2010; 31 (2). Available from: https://doi.org/10.1002/jcc.21334
12. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, <I>et al</I>. The Protein Data Bank. Nucleic Acids Research. 2000; 28 (1). Available from: https://doi.org/10.1093/nar/28.1.235
13. Morris GM, Lim-Wilby M. Molecular docking. In: Kukol A, editor. Molecular Modeling of Proteins.. 2008; Available from: https://doi.org/10.1007/978-1-59745-177-2_19
14. Laskowski RA, Swindells MB. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. Journal of Chemical Information and Modeling. 2011; 51 (10). Available from: https://doi.org/10.1021/ci200227u
15. Lipinski CA. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discovery Today: Technologies. 2004; 1 (4). Available from: https://doi.org/10.1016/j.ddtec.2004.11.007
16. Huang SY, Zou X. Advances and Challenges in Protein-Ligand Docking. International Journal of Molecular Sciences. 2010; 11 (8). Available from: https://doi.org/10.3390/ijms11083016
17. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, <I>et al</I>. UCSF Chimera—A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004; 25 (13). Available from: https://doi.org/10.1002/jcc.20084
18. Yang J, Roy A, Zhang Y. Protein–ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics. 2013; 29 (20). Available from: https://doi.org/10.1093/bioinformatics/btt447
19. McGann M. FRED and HYBRID docking performance on standardized datasets. Journal of Computer-Aided Molecular Design. 2012; 26 (8). Available from: https://doi.org/10.1007/s10822-012-9584-8
20. Kitchens CA, Wang X, Salsbury FR. Molecular docking: A case study of the effects of ligand conformation on binding interactions. <I>Journal of Molecular Modeling</I>. 2011;17(2):309–17.
21. Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, <I>et al</I>. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry. 1998; 19 (14). Available from: https://doi.org/10.1002/(sici)1096-987x(19981115)19:14<1639::aid-jcc10>3.0.co;2-b
22. Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein–ligand docking using GOLD. Proteins: Structure, Function, and Bioinformatics. 2003; 52 (4). Available from: https://doi.org/10.1002/prot.10465
23. Irwin JJ, Shoichet BK. ZINC − A Free Database of Commercially Available Compounds for Virtual Screening. Journal of Chemical Information and Modeling. 2005; 45 (1). Available from: https://doi.org/10.1021/ci049714+
24. Li X, Zhang H. Molecular docking, Challenges and opportunities for drug discovery. <I>Future Medicinal Chemistry</I>. 2019, 11(14), 1741–1744.
25. Sterling T, Irwin JJ. ZINC 15 – Ligand Discovery for Everyone. Journal of Chemical Information and Modeling. 2015; 55 (11). Available from: https://doi.org/10.1021/acs.jcim.5b00559
26. Barakat KH, Gajendrarao P, Tuszynski JA, Houghton M. Molecular docking and dynamic simulations for drug discovery. <I>Expert Opinion on Drug Discovery</I>. 2012, 7(11), 1039–1055.
27. Liu J, Wang R, Lu Y. Protein-ligand interaction studies using docking and molecular dynamics simulations. Current Topics in Medicinal Chemistry. 2017; 17 (24). Available from: https://doi.org/10.1021/acs.jcim.0c00057.s001
28. Sanner MF. Python, A programming language for software integration and development. <I>Journal of Molecular Graphics and Modelling</I>. 1999, 17(1), 57–61.
29. Ercan S, Çınar E. A molecular docking study of potential inhibitors and repurposed drugs against SARS-CoV-2 main protease enzyme. Journal of the Indian Chemical Society. 2021; 98 (3). Available from: https://doi.org/10.1016/j.jics.2021.100041
30. Banerjee R, Perera L, Tillekeratne LMV. Potential SARS-CoV-2 main protease inhibitors. Drug Discovery Today. 2021; 26 (3). Available from: https://doi.org/10.1016/j.drudis.2020.12.005
31. Ansari JA, Ahmad MK, Fatima N, Azad I, Mahdi AA, Satyanarayan GNV, <I>et al</I>. Chemical Characterization, <i>In-silico</i> Evaluation, and Molecular Docking Analysis of Antiproliferative Compounds Isolated from the Bark of <i>Anthocephalus cadamba</i> Miq.. Anti-Cancer Agents in Medicinal Chemistry. 2022; 22 (20). Available from: https://doi.org/10.2174/1871520622666220204123348
32. Hu Q, Xiong Y, Zhu GH, Zhang YN, Zhang YW, Huang P, <I>et al</I>. The SARS‐CoV‐2 main protease (M<sup>pro</sup>): Structure, function, and emerging therapies for COVID‐19. MedComm. 2022; 3 (3). Available from: https://doi.org/10.1002/mco2.151
33. Shivanika C, Deepak Kumar S, Ragunathan, V, Tiwari P, Sumitha A, Brindha Devi P. Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. Journal of Biomolecular Structure and Dynamics. 2022; 40 (2). Available from: https://doi.org/10.1080/07391102.2020.1815584
34. Mohammed FS, Pehlivan M, Sevindik E, Akgul H, Sevindik M, Bozgeyik I, <I>et al</I>. Pharmacological properties of edible Asparagus acutifolius and Asparagus officinalis collected from North Iraq and Turkey (Hatay). Acta Alimentaria. 2021; 50 (1). Available from: https://doi.org/10.1556/066.2020.00204
35. Gajendra S, Chakraborty R, Bhattacharya S. Virtual screening of bioactive components from <I>Anthocephalus cadamba</I> targeting SARS-CoV-2 main protease. <I>Journal of Biomolecular Structure and Dynamics</I>. 2021, 39(17), 6240–6255.
36. Chakravarthi G, Remidicherla SS, Malothu N. A validated LC-MS/MS method for detection of nitrosamine impurities in favipiravir API. <I>Indian Journal of Pharmaceutical Education and Research</I>. 2025, 59(1s), S256–S263.
37. Veerareddy V, Kumaraswamy G. Development and Validation of a New RP-HPLC Method for the Simultaneous Estimation of Nirmatrelvir, Ritonavir and Molnupiravir in Formulated Nanosponges, Plasma Samples and its Pharmacokinetic Study. Indian Journal of Pharmaceutical Education and Research. 2024; 58 (4). Available from: https://doi.org/10.5530/ijper.58.4.142
38. Swetha SR, Pravallika S, Chakravarthi G. Cleaning method development and validation by UV method for quantitative assessment of favipiravir residue in manufacturing area. Annals of Phytomedicine: An International Journal. 2022; 11 (Special Issue 1). Available from: https://doi.org/10.54085/ap.trips.2022.11.1.15
39. Remidicherla SS, Chakravarthi G, Madhavi A, Malothu N. DEVELOPMENT AND VALIDATION OF AN LC-MS/MS METHOD FOR NITROSAMINE IMPURITY DETECTION IN TAMSULOSIN HYDROCHLORIDE. International Journal of Applied Pharmaceutics. 2025; 17 (2). Available from: https://doi.org/10.22159/ijap.2025v17i2.52810
40. Asha BR, Goudanavar P, Koteswara Rao GSN, Kumaraswamy G. QbD driven targeted pulmonary delivery of dexamethasone-loaded chitosan microspheres: Biodistribution and pharmacokinetic study. Saudi Pharmaceutical Journal. 2023; 31 (9). Available from: https://doi.org/10.1016/j.jsps.2023.101711
41. Hu Q, Xiong Y, Zhu GH, Zhang YN, Zhang YW, Huang P, <I>et al</I>. The SARS‐CoV‐2 main protease (M<sup>pro</sup>): Structure, function, and emerging therapies for COVID‐19. MedComm. 2022; 3 (3). Available from: https://doi.org/10.1002/mco2.151
© 2026 Published by Krupanidhi College of Pharmacy. This is an open-access article under the CC BY-NC-ND license (https://creativecommons.org/licenses/by-nc-nd/4.0/)
Subscribe now for latest articles and news.


